Structural Modeling of MHC-bound Antigen
Peptides
|
|
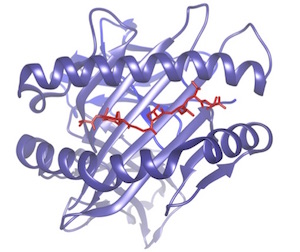
In the context of cancer immunotherapy it
is key to be able to identify which peptides
specific to the tumor may bind to the MHC to
be presented to the immune system
(neoantigens). Even more important is to
predict which of these neoantigens will
trigger a significant T-cell response.
While sequence-based predictors show some
success for MHC binding, predicting
antigenicity remains challenging without
structural information. In collaboration with
V. Zoete at SIB and CHUV, we use a combination
of docking, protein modeling and molecular
dynamics simulations to model the structure
and dynamics of MHC-bound peptides. A reliable
structural model will be the stepping stone to
identify determinants of TCR binding, and thus
of antigenicity.
|
|
In Situ Data Analytics for Next Generation
Molecular Dynamics Workflows
|
Next-generation
high-performance computing (HPC) systems will
have dramatically larger compute performance
than current systems do, which translastes
into the ability to create huge amounts of MD
data. But since storage systems will not
increase their bandwidth concommittently, data
storage and analysis will become the main
bottleneck. The
Analytics4MD project tackles the data
challenges of MD simulations at the exascale
through (1) creation of novel data analytics
algorithms ideal for in situ data analysis of
relevant structural molecular properties, (2)
definition of MD-based machine learning (ML)
techniques to automatically identify the
molecular domains where the properties reside
at runtime, and (3) integration of both
algorithms and techniques into MD workflows at
the extreme scale.
Analytics4MD is a collaborative effort between
the University of Tennessee Knoxville, the
University of Southern California, the
University of New Mexico, and Weill Cornell
Medicine, funded by an combined grant of the
American National Science Foundation. |
 |
|
The T Cell Receptor as a Mechanosensor
|
|
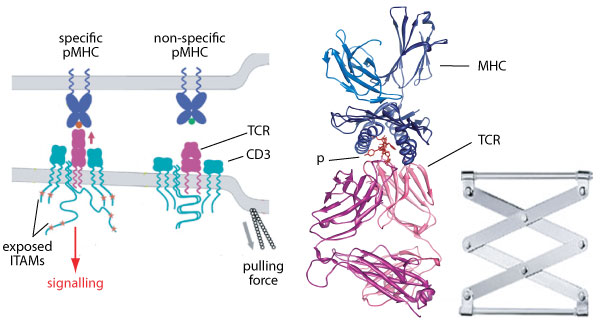
T lymphocyte response is determined by the
interaction of the T cell receptor (TCR) with
peptide (p) antigens presented by MHC
molecules. While he molecular determinants of
recognition at the TCR-pMHC interface have
been extensively studied, the mechanisms by
which the activation signal is transmitted
into the T cell are still to be discovered.
Understanding and optimizing TCR signaling is
key to the development of therapeutic
approaches such as adoptive transfer of
genetically engineered T cells for cancer
immunotherapy. It has recently emerged that
the TCR functions as a mechanosensor that
recognizes agonist antigens when subjected to
cytoskeletal forces. However, the
molecular mechanisms underpinning the
mechanosensor function remain unknown. We use
steered molecular dynamics to study the effect
of external forces on the TCR bound to agonist
or non-agnonist antigens. As nonequilibrium
simulations require many replica, this project
involves large-scale computational resources,
such as the Piz Daint supercomputer at the Swiss
National Supercomputing Centre. |
|
Quantifying Allostery in Proteins: the
Thermodynamic Coupling Function Analysis
Method
|
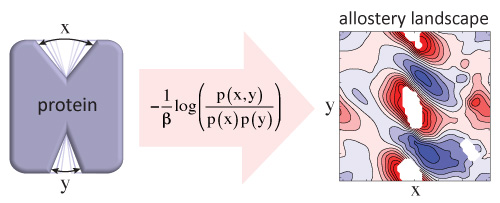
Going beyond the usual two-state models of
allostery, in collaboration with M. LeVine and
H- Weinstein at WCM, we introduced a
statistical mechanical formalism to describe
rigorously the coupling between two collective
variables that represent key conformational
changes in a protein, such as ligand binding
and activation. We show that allosteric
coupling is best represented as a
two-dimensional thermodynamic coupling
function (TCF).
We combined the TCF formalism and a Markov
state model analysis of the human dopamine
transporter (hDAT), revealing a non-trivial
thermodynamic coupling landscape between the
sodium release and intracellular gating steps.
We make available online AlloDeco,
a fast version of the TCF analysis to
decompose allosteric coupling based on
coarse-grained Gaussian Network models.
|
|
|
Neurotransmitter Sodium-coupled
Transporter Proteins
|
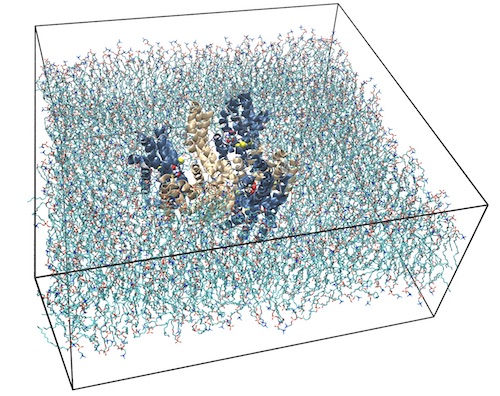 |
The glutamate transporter GltPh is a homolog
of mammalian excitatory amino acid
transporters (EAATs) that mediate glutamate
re-uptake after discharge at the neuronal
synaptic cleft. In the transport cycle, the
three homotrimeric transport domains (blue)
undergo an elevator-like motion relative to a
scaffold domain (wheat). With a team at Weill
Cornell, we reported
in
Nature significantly higher transport
rates for a GltPh construct in which key
residues were mutated to mimic human EAAT1.
The mutant adopted a novel “unlocked”
conformation (PDB 4X2S)
that for which MD simulations showed to be
stable only if hydrophobic molecules such as
lipid tails insert between domains.
We are currently expanding this work to
quantify the contributions of single residues
to the stability of several intermediate
states. In particular, we are interested in
understancing the allosteric coupling between
the closure of the ligand binding site by the
HP2 loop (the "elevator door") and the inward
elevator motion. This coupling that prevents
transport of sodium without substrate ("sodium
leak"), is essential to the symport function.
We are also investigating the role of
protonation of key acidic residues in the
ligand binding site.
|
|
Past Research
Please go to the next
page
|