Assessment of force fields on small
peptide conformational equilibria
|
| In
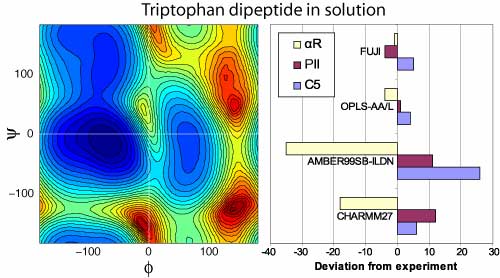
this project with Alex Tzanov at NYU, we used
the driven adiabatic free energy dynamics
method to efficiently sample the
conformational space of small peptides whose
conformational preferences have been
experimentally measured, both in solution and
in the gas phase. We computed the free energy
surface of these peptides in the space of key
dihedral angles and for which there are
experimental results for |
 |
|
Unified free energy dynamics (UFED)
|
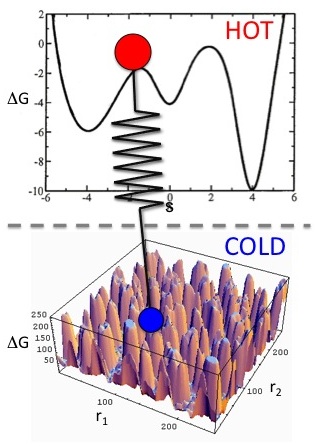 |
Driven adiabatic free energy dynamics
(dAFED, also known as TAMD) is an efficient
approach for the sampling of conformational
equilibria in complex systems and the
generation of associated free energy
hypersurfaces in terms of a set of collective
variables. The method has recently been
generalized to exploit the strengths of other
methods, namely using Gaussian-based adaptive
bias potentials to disfavor hitherto visited
regions of configuration space and using the
thermodynamic force instead of the probability
density to reconstruct the free energy
surface. The unified free energy dynamics
(UFED) scheme is shown to outperform both
metadynamics and adiabatic free energy
dynamics in several example cases including
the alanine dipeptide and the met-enkephalin
oligopeptide. In addition, UFED is not limited
to one- or two-dimensional collective
variables. We have showed the applicability of
the method to construct free energy
hypersurfaces as a function of up to 6
dihedral angles.
Both dAFED and UFED methods are
implemented within an older version of the PLUMED
plugin, which works with a large number of
molecular dynamics software packages. The source code is available
here,
and the associated manual pages here.
A Matlab package to reconstruct free energy surfaces from the average force, from metadynamics-like hills, or from reweighted histograms is also available here.
|
|
Alchemical free energy differences with
enhanced conformational sampling
|
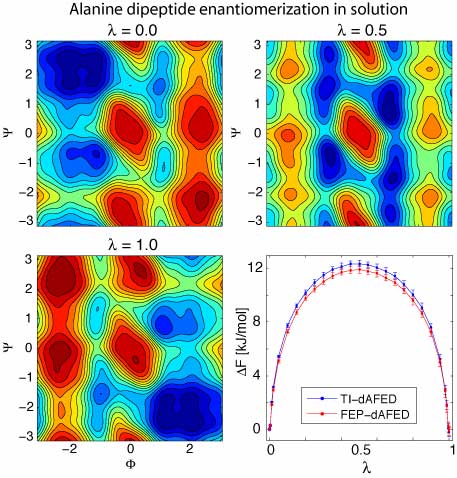
Alchemical free energy simulations are
commonly used to calculate relative binding or
solvation free energies in molecular systems.
The convergence of alchemical free energy
calculations is often hampered by inefficient
sampling of the conformational degrees of
freedom, which remain trapped in metastable
substates. We have recently shown that
thermodynamic integration (TI) or free energy
perturbation (FEP) can be combined with the
driven adiabatic free energy dynamics (dAFED)
method, in order to enhance conformational
sampling along a set of chosen collective
variables. The resulting TI-dAFED or FEP-dAFED
methods have been validated on a
two-dimensional analytical problem as well as
by calculating the enantiomerization free
energy of the alanine dipeptide in explicit
solvent.
|
 |
|
Force field integration
|
 |
In 2010, I teamed up with the Gromacs
developers to integrate Charmm27, one of the
most popular force fields, to the Gromacs
molecular dynamics package. The new
implementation was rigorously tested on
protein and nucleic acid systems.
In parallel, together with Vincent Zoete at
the Swiss Institute of Bioinformatics, we
developed the SwissParam
web server. For any small molecule, this
service provides quick generation of topology
and force field files that can be used
directly within the Charmm and Gromacs
softwares.
|
|
Development of peptide inhibitors for MAP
kinases
|
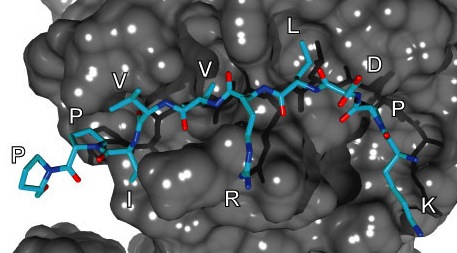
| In a
collaboration between the Molecular Modeling
Group at the Swiss Institute of Bioinformatics
and the (now defunct) pharmaceutical company
Xigen, we developed peptide inhibitors for
several MAP kinases. Using binding free energy
decomposition methods (and chemical
intuition), we proposed peptides including
peptidase-resistant D-amino acids. These
peptides were then synthesized and tested in
vitro by Xigen, and further optimized through
several rounds of modelization and
measurements. Note that these results were
never published due to intellectual property
issues. |
 |
|
T cell receptor / peptide-MHC interactions
|
 |
The group of Olivier Michielin has an
ongoing interest in understanding the
mechanisms underlying T cell activation
through T cell receptor (TCR) binding to
specific peptide-MHC proteins on the surface
of antigen-presenting cells. As part of this
effort, we conducted an extensive steered
molecular dynamics study of three related
TCR-pMHC complexes.
In a first part of this study we developed the
individual steering scheme in which proteins
are pulled apart while their overall structure
is preserved. We then tried to apply the
Jarzynski identity to calculate absolute
binding free energies for these TCR-pMHC
complexes, based on a large number of slow
nonequilibrium unbinding trajectories. The
results illustrated the now well-recognized
convergence difficulties of the Jarzynski
identity for processes with large activation
free energies.
In the second part of this study, we took
advantage extremely large steered molecular
dynamcics data sets to calculate average
values of several observables as a function of
the TCR-pMHC distance. These observables
include energetic components, number of
H-bonds or hydrophobic contacts and the
localization of trapped water molecules. |
|
Nonequilibrium statistical mechanics
|
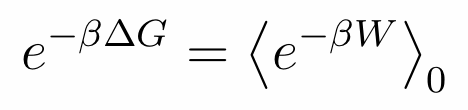
|
My Ph.D thesis at ETH Zurich focused on two
complementary aspects of molecular dynamics:
thermostating and nonequilibrium simulation.
In particular, the Jarzynski identity states
that the equilibrium free energy of a process
can be reconstructed by averaging the external
work performed in many nonequilibrium
realizations of the process. In such
nonequilibrium simulations, the excess heat is
extracted from the system by the thermostat
used to generate the desired thermodynamical
ensemble. The central theoretical results was
a proof of the Jarzynski identity based on the
specific equations of motion used in
thermostated molecular dynamics, without any
further assumptions.
Here you can download my PhD
thesis. |
|